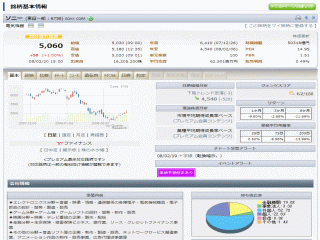
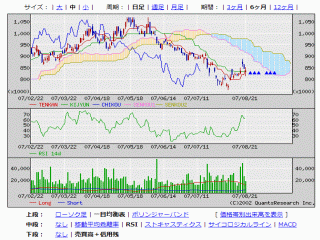
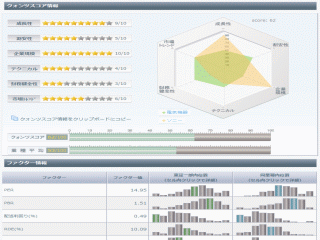
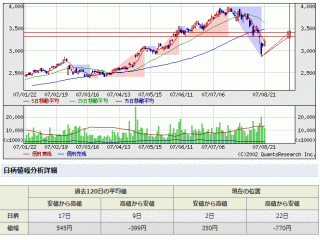
選択されたページは表示期間が切れたため、表示することができません。それ以外のTDnet適時開示情報はこちらへ
株マップ.comへ(登録無料)
デイトレ株マップ.comこちらへ Hot!!!
リアルタイム株価に対応、さらに充実した分析コンテンツが準備されています。